

Implementación de programas de transición niño-adulto para pacientes con cardiopatías congénitas
28 agosto, 2022 - 8 min de lectura

Versión 1 - 28/08/22
Debido a la falta de reconocimiento oportuno y de continuidad del cuidado en los casos intervenidos, la mayoría de los pacientes con cardiopatías congénitas en la infancia siguen teniendo un riesgo cardiovascular moderado-alto el resto de la vida. Por tanto, se recomienda diseñar e implementar programas institucionales de transición para pacientes con cardiopatías congénitas que favorezcan su educación, el seguimiento médico continuo y una transición adecuada de un grupo clínico pediátrico a uno de adultos.
Certeza de la evidencia: baja por imprecisión, variabilidad metodológica y heterogeneidad clínica y estadística entre los estudios primarios.
Otro mensaje clave:
- La presencia de grupos interdisciplinarios exclusivos para la atención de pacientes con cardiopatías congénitas favorece el éxito de los programas de transición.
Es importante saber lo que no se conoce
• Se desconoce la incidencia y la prevalencia de cardiopatías congénitas que alcanzan la edad adulta en nuestro país.
• Dada la inexistencia de programas de transición niño-adulto en nuestro país, se desconoce el impacto y la magnitud de la pérdida de seguimiento de los pacientes con cardiopatías congénitas.
Antecedentes
Las cardiopatías congénitas (CC) son un grupo de enfermedades producto de alteraciones anatómicas que modifican el funcionamiento normal del sistema cardiovascular (1). Algunas se manifiestan desde el nacimiento llegando incluso a poner en riesgo la vida del recién nacido (2), mientras que otras pueden pasar desapercibidas durante toda la vida, o presentar síntomas hasta la adultez (3). En la actualidad, 9 de cada 1000 nacidos vivos presenta algún tipo de CC (4). Los avances médicos de las últimas décadas han aumentado la sobrevida de estos pacientes, que llega a ser del 90% en países desarrollados (5).
Este aumento en la expectativa de vida implica la necesidad de seguimiento especializado continuo. Aún después de intervenido el defecto anatómico, estos pacientes pueden presentar defectos residuales que aumentan el riesgo de diversos problemas cardiovasculares futuros. Se estima que alrededor del 55% de estos pacientes tienen un riesgo cardiovascular residual al menos moderado, definido como muerte prematura, necesidad de re intervención u otras complicaciones cardiovasculares mayores (6).
Normalmente estos pacientes son acompañados durante la primera parte de su vida por el equipo de atención pediátrica y como adultos su seguimiento debería estar a cargo del equipo de atención al adulto. Sin embargo, es frecuente que algunos pacientes pierdan el seguimiento durante esta transición, bien sea por factores del paciente o del sistema (7, 8). La importancia de garantizar la continuidad de la atención se sustenta en la capacidad que tienen los centros especializados en CC de disminuir la mortalidad comparado con centros con menos experiencia (9).
En Bogotá 15,1 de cada 10.000 recién nacidos vivos presentan algún tipo de CC (10), según el Programa de Vigilancia y Seguimiento de Niños con Anomalías Congénitas de la Secretaría de Salud. Por otra parte, en Colombia se estima que 150 niños por cada 1’000.000 de habitantes requerirá algún tipo de cirugía cardíaca, sin embargo, solo un tercio de los pacientes son intervenidos (11). Esto implica que la proporción de pacientes con CC que alcanza la adolescencia o la adultez con persistencia del defecto anatómico congénito, vivirá las consecuencias sobre su salud y calidad de vida que esto pueda significar en cada caso. Como resultado de esta situación se desconoce la prevalencia de CC en adultos en nuestro país.
En este contexto, el propósito de este Recado es reseñar la revisión sistemática con metanaálisis de Moons y colaboradores (12), que explora puntos de intervención para optimizar la transición en el seguimiento de la adolescencia a la adultez temprana.
Información en la que se basa este Recado
El estudio de Moons y colaboradores es una revisión sistemática con metaanálisis que incluyó 17 estudios con un total de 6847 pacientes; 10 estudios de Estados Unidos (EU), 5 de Canadá, 1 de Bélgica y 1 de Suecia. Uno de los estudios incluidos fue un ensayo clínico aleatorizado con educación sobre la enfermedad como intervención y el seguimiento médico como desenlace (13). De estos estudios, 13 recolectaron pacientes de centros de atención pediátrica, los otros 4 reclutaron pacientes de centros de atención de adultos.
Aunque los autores identificaron gran variabilidad metodológica entre los estudios, definieron tres formas de falta de seguimiento:
- Más de 2-3 años entre consultas al equipo especializado en cardiopatías congénitas.
- No tener visitas a cardiología por más de 4-5 años después de pasar al grupo de atención del adulto.
- Más de 12 años sin visitas al cardiólogo entre la adolescencia y la adultez.
Resultados
La proporción de pacientes que abandonan el seguimiento se presenta en la siguiente tabla. El estimado agregado de los estudios fue 26.1% (IC95% 19.2-34.6, I2 97%) con un rango de 3.6-62.7%.
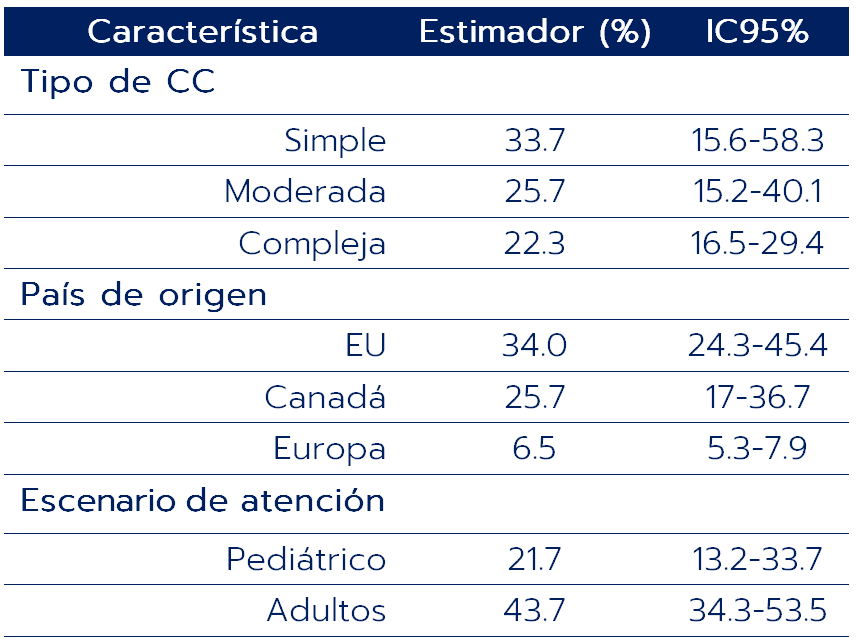
En el análisis de sensibilidad se mantuvieron las diferencias entre haber sido reclutado en un centro pediátrico vs adulto, entre estar ubicado en EU o Canadá vs Europa y entre tener CC simple o moderada vs compleja.
Los autores plantean como explicación a las diferencias entre la efectividad de los programas de transición de EU, Canadá y Europa aspectos como el acceso universal a la salud; la aplicación de políticas institucionales en los centros europeos para transferir sistemáticamente los pacientes pediátricos a los centros de cuidado adulto (efectividad 85%); la presencia de un mayor número de centros y personal dedicados exclusivamente al manejo de este tipo de pacientes; o la complejidad de los tipos de CC incluidas en los programas de seguimiento (aquellos con enfermedad más grave tienen mayor frecuencia de visitas médicas).
Información sobre la evidencia que soporta este Recado
El estudio es de calidad moderada de acuerdo con la herramienta AMSTAR 2. Los autores consideraron que los estudios primarios tuvieron una calidad global moderada o alta de acuerdo con Newcastle-Ottawa Scale, tampoco detectaron riesgo de sesgo de publicación.
Referencias
Yamagishi H. Clinical Developmental Cardiology for Understanding Etiology of Congenital Heart Disease. J Clin Med. 2022 Apr 24;11(9):2381. doi: 10.3390/jcm11092381. PMID: 35566507; PMCID: PMC9104584.
Mandalenakis Z, Rosengren A, Skoglund K, Lappas G, Eriksson P, Dellborg M. Survivorship in children and young adults with congenital heart disease in Sweden. JAMA Intern Med. 2017;177:224–230. DOI: 10.1001/jamainternmed.2016.7765.
Iversen K, Vejlstrup NG, Sondergaard L, Nielsen OW. Screening of adults with congenital cardiac disease lost for follow‐up. Cardiol Young. 2007;17:601–608. DOI: 10.1017/S1047951107001436
Sandoval N. Congenital heart disease in Colombia and worldwide. Rev Colomb Cardiol. 2015;22(1):e1–e2.
Ávila P, Mercier LA, Dore A, Marcotte F, Mongeon FP, Ibrahim R, Asgar A, Miro J, Andelfinger G, Mondésert B, de Guise P, Poirier N, Khairy P. Adult congenital heart disease: a growing epidemic. Can J Cardiol. 2014 Dec;30(12 Suppl):S410-9. doi: 10.1016/j.cjca.2014.07.749. Epub 2014 Sep 28. PMID: 25432136.
Warnes CA, Liberthson R, Danielson GK, Dore A, Harris L, Hoffman JI, Somerville J, Williams RG, Webb GD. Task force 1: the changing profile of congenital heart disease in adult life. J Am Coll Cardiol. 2001 Apr;37(5):1170-5. doi: 10.1016/s0735-1097(01)01272-4. PMID: 11300418.
Moons P, Hilderson D, Van Deyk K. Implementation of transition programs can prevent another lost generation of patients with congenital heart disease. Eur J Cardiovasc Nurs. 2008 Dec;7(4):259-63. doi: 10.1016/j.ejcnurse.2008.10.001. PMID: 19013410.
Fernandes SM, Verstappen A, Clair M, Rummell M, Barber D, Ackerman K, Dummer K, Mares JC, Cannobio MM, Reardon LC, Long J, Crumb S, Bhatt A, Takahashi M, Khairy P, Williams R, Landzberg MJ, Moe T, Pearson D; Adult Congenital Heart Association and the Adult Congenital Cardiac Care Associate Research Group. Knowledge of Life-Long Cardiac Care by Adolescents and Young Adults with Congenital Heart Disease. Pediatr Cardiol. 2019 Oct;40(7):1439-1444. doi: 10.1007/s00246-019-02154-8. Epub 2019 Jul 31. PMID: 31367952.
Mylotte D, Pilote L, Ionescu-Ittu R, Abrahamowicz M, Khairy P, Therrien J, Mackie AS, Marelli A. Specialized adult congenital heart disease care: the impact of policy on mortality. Circulation. 2014 May 6;129(18):1804-12. doi: 10.1161/CIRCULATIONAHA.113.005817. Epub 2014 Mar 3. PMID: 24589851.
Tassinari S, Martínez-Vernaza S, Erazo-Morera N, Pinzón-Arciniegas MC, Gracia G, Zarante I. Epidemiología de las cardiopatías congénitas en Bogotá, Colombia en el período comprendido entre 2001 y 2014: ¿Mejoría en la vigilancia o aumento en la prevalencia. biomedica [Internet]. 1 de mayo de 2018 [citado 13 de agosto de 2022];38(Sup1):141-8. Disponible en: https://revistabiomedica.org/index.php/biomedica/article/view/3381
Sandoval N. Congenital heart disease in Colombia and Worldwide. Rev Col Cardiol. 2015;22:1-2. https://doi.org/10.1016/j.rccar.2015.03.005
Moons P, Skogby S, Bratt EL, Zühlke L, Marelli A, Goossens E. Discontinuity of Cardiac Follow-Up in Young People With Congenital Heart Disease Transitioning to Adulthood: A Systematic Review and Meta-Analysis. J Am Heart Assoc. 2021 Mar 16;10(6):e019552. doi: 10.1161/JAHA.120.019552. Epub 2021 Mar 4. PMID: 33660532; PMCID: PMC8174191.





